CFTR: Products
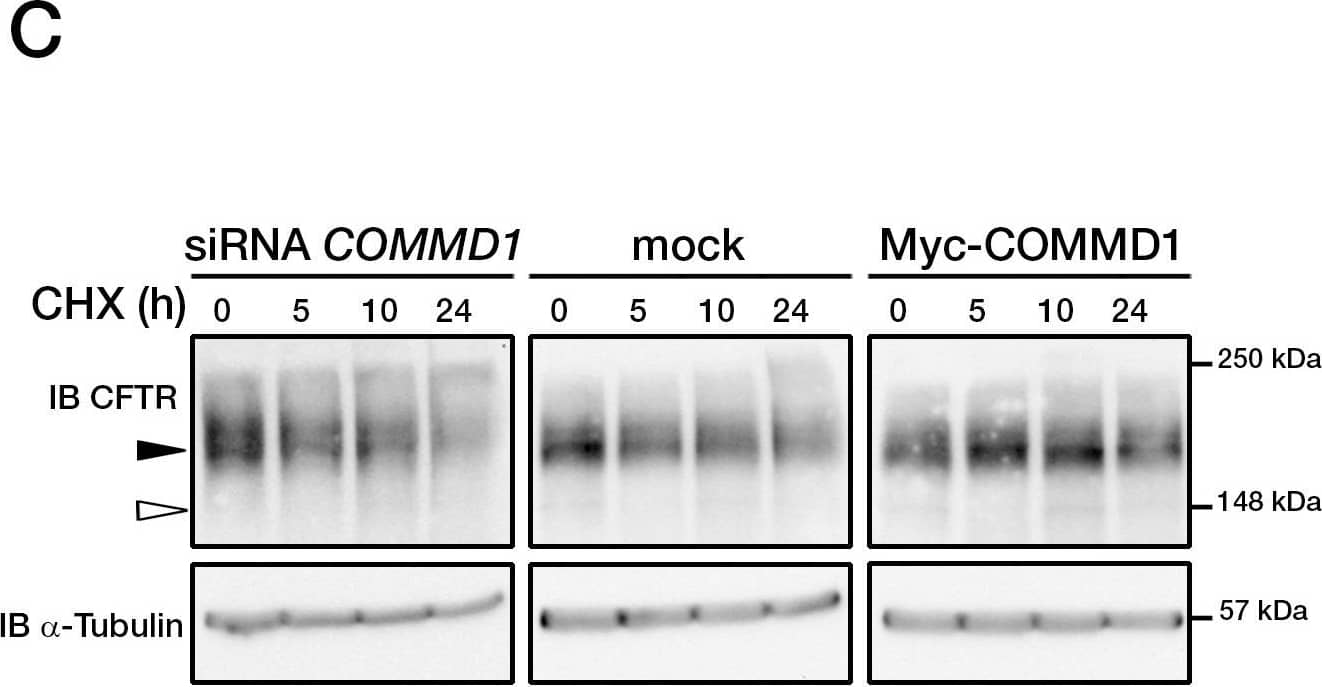
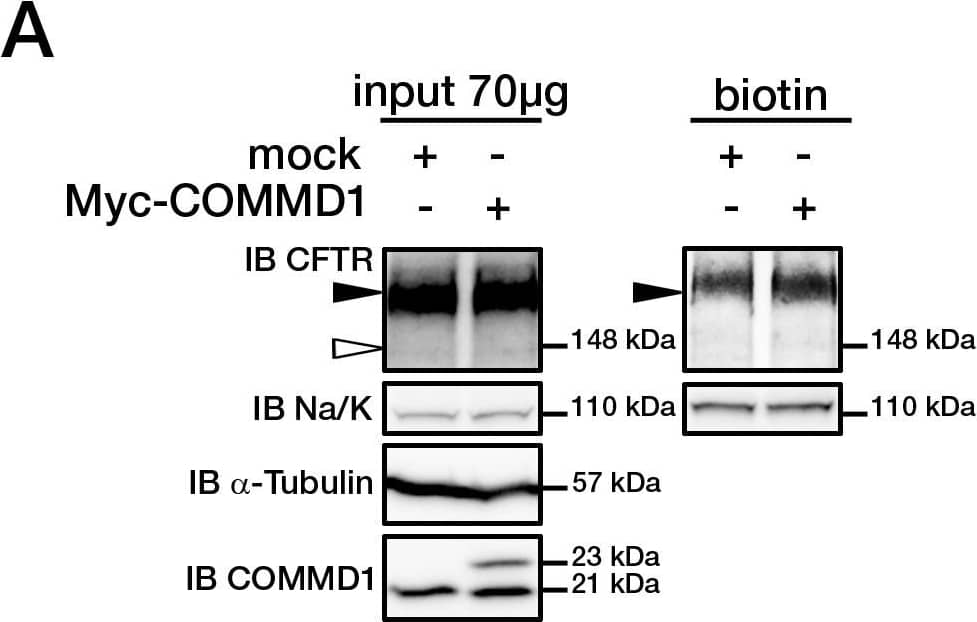
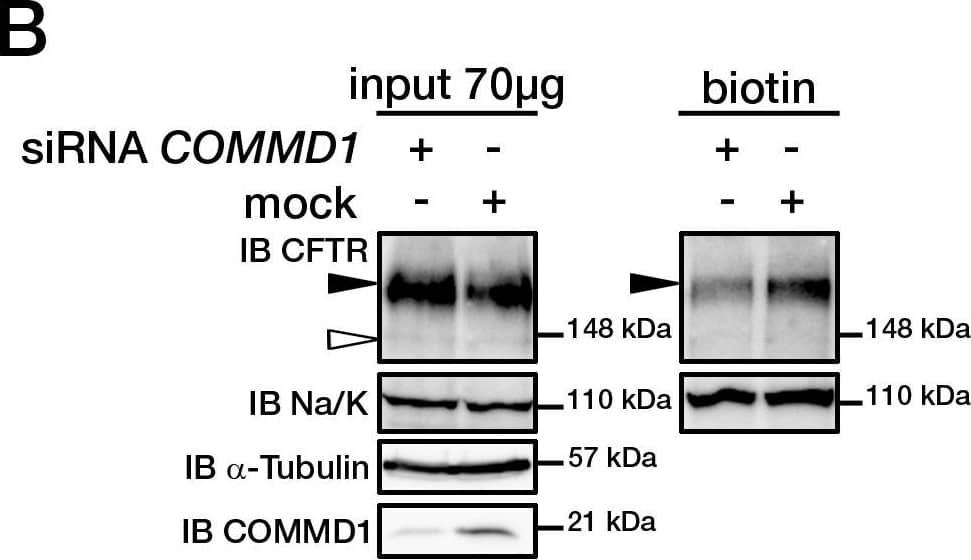
Cystic fibrosis transmembrane conductance regulator (CFTR) is a multi-pass transmembrane protein that functions as a chloride channel. CFTR belongs to the ATP-binding cassette (ABC) superfamily. Mutations in CFTR cause the pulmonary disease, cystic fibrosis (CF). Specifically, deletion of phenyalanine at position 508 (DeltaF508-CFTR) results in a folding defect which impairs chloride channel function. The mechanism by which channel dysfunction relates to disease symptoms is a focus of intense research. CFTR dysfunction results in disruption of ion transport and subsequent blockage of airways by secreted mucus. CFTR may also play a role in the skeletal muscle atrophy and dysfunction that characterizes CF. In addition, CFTR-mediated chloride secretion underlies fluid accumulation and cyst growth in autosomal dominant polycystic kidney disease (ADPKD).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}